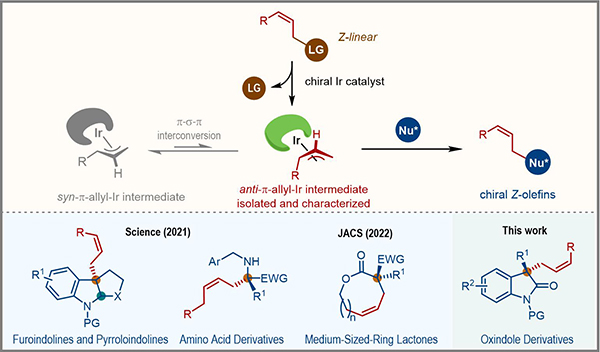
Z-烯烴片段廣泛存在于天然產物和藥物分子中�。但由于大位阻取代基位于雙鍵同側����,Z-烯烴相對于E-烯烴熱力學不穩(wěn)定,因此其高選擇性合成極具挑戰(zhàn)性��。過渡金屬催化的不對稱烯丙基取代反應通過利用親核試劑捕獲π-烯丙基金屬配合物中間體��,可以高效構建含有烯烴片段的手性化合物��。但是該類反應一般經(jīng)歷熱力學穩(wěn)定的syn-π-烯丙基金屬配合物�����,得到末端烯烴或者E-烯烴產物�����。
2021年�,中國科學院上海有機化學研究所游書力研究團隊利用“活潑前手性親核試劑捕獲亞穩(wěn)態(tài)anti-π-烯丙基金屬配合物”的策略,實現(xiàn)了銥催化Z式保留不對稱烯丙基取代反應���,高效地構建了一系列含有Z-烯烴片段的復雜手性分子(圖1����,Science 2021, 371, 380; J. Am. Chem. Soc. 2022, 144, 4770.)。在這類反應中��,anti-π-烯丙基銥配合物一般被推測為關鍵的催化活性中間體�����,對其分離�、表征及性質研究具有重要的意義。但該中間體熱力學不穩(wěn)定�,容易通過π-σ-π異構化過程轉化為熱力學穩(wěn)定的syn-π-烯丙基銥配合物,導致其分離表征十分具有挑戰(zhàn)性���。
圖1 銥催化Z式保留不對稱烯丙基取代反應
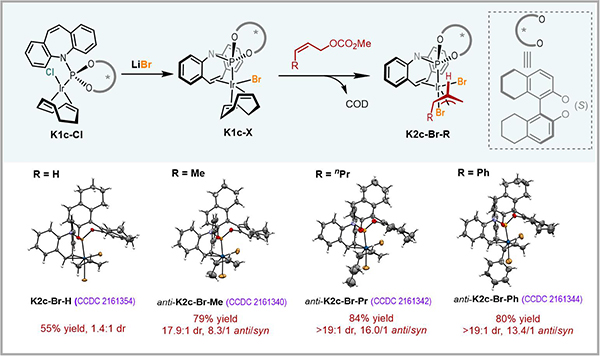
在前期工作中�����,研究人員通過核磁共振磷譜(31P NMR)和高分辨質譜(HRMS)對一類手性磷/烯烴配體衍生的anti-π-烯丙基銥配合物(三氟甲磺酸根為抗衡陰離子)的生成以及異構化過程進行了表征�����,但未能實現(xiàn)該配合物的分離鑒定�。最近研究人員通過向體系中引入強配位的鹵離子,提升anti-π-烯丙基銥配合物的穩(wěn)定性����,成功實現(xiàn)了一系列anti-π-烯丙基銥配合物的合成,并通過單晶X射線衍射確證了其結構(圖2)�。同時通過核磁共振磷譜表征了anti-π-烯丙基銥配合物向熱力學穩(wěn)定的syn-π-烯丙基銥配合物的異構化過程,并且證實異構化所需的時間長于親核進攻����。這是實現(xiàn)Z式保留的不對稱烯丙基取代反應的關鍵因素���。
圖2 anti-π-烯丙基銥配合物的合成及表征
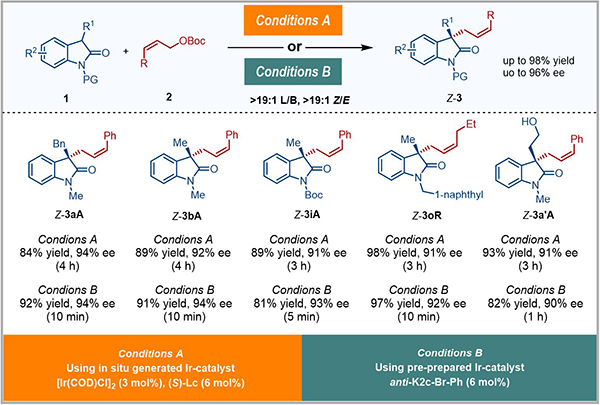
該類anti-π-烯丙基銥配合物可以高效催化一系列吲哚-2-酮衍生的前手性親核試劑與Z-烯丙基碳酸酯的Z式保留的不對稱烯丙基取代反應(圖3)��。研究發(fā)現(xiàn)使用預先制備的銥配合物能取得與原位生成的銥催化劑相當?shù)氖章剩?1~97%)和選擇性(L/B > 19/1, Z/E >19/1, 90-94% ee)�����,并將反應時間從2小時到1天���,縮短為5分鐘到1小時。
圖3 銥催化吲哚-2-酮衍生物參與的Z式保留不對稱烯丙基取代反應
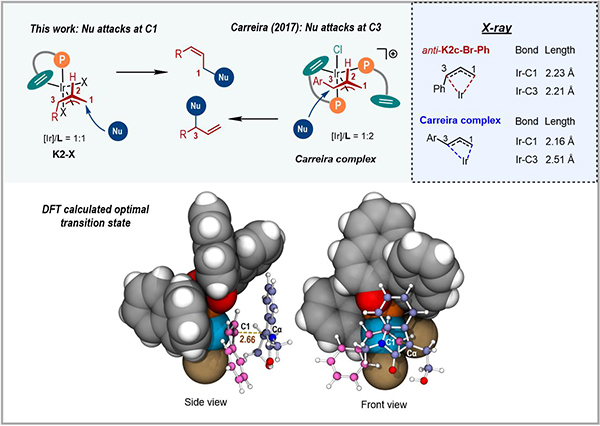
研究人員通過分析anti-π-烯丙基銥配合物([Ir]/L = 1:1)的幾何和電子結構揭示了反應區(qū)域選擇性的成因(圖4)����。由于磷配體并不處于烯丙基任何一端(C1和C3)的反位,使得C1–Ir鍵和C3–Ir鍵的鍵長以及Mayer鍵級基本相同,因此親核試劑優(yōu)先進攻位阻較小的C1位����。這與文獻報道的由同類手性配體衍生的銥配合物([Ir]/L = 1:2)明顯不同。研究人員進一步使用DFT計算考察了吲哚-2-酮負離子進攻anti-π-烯丙基銥配合物的過渡態(tài)����,提出了該反應的手性控制模型。
圖4 銥催化Z式保留不對稱烯丙基取代反應的區(qū)域/對映選擇性模型
綜上所述��,該項工作報道了Z式保留不對稱烯丙基取代反應中關鍵的anti-π-烯丙基銥配合物中間體的合成����、分離與表征,深入研究了其形成���、異構化和與親核試劑反應的機制�。在此基礎上發(fā)展了一類新型的Z式保留不對稱烯丙基取代反應��,提出了反應區(qū)域選擇性成因和手性誘導模型����。研究論文以全文形式在線發(fā)表于Nature Catalysis(DOI: 10.1038/s41929-022-00879-z)。這項成果對于進一步發(fā)展Z式保留不對稱烯丙基取代反應���,合成手性Z-烯烴奠定了堅實的基礎��。
上述研究工作得到了科技部���、國家自然科學基金委����、中國科學院�����、上海市科委和騰訊基金會的資助�����。